



La Neuropatía óptica hereditaria de Leber (NOHL) es una entidad infrecuente y con pronóstico visual sombrío. En pacientes monoculares un alto índice de sospecha es necesario para establecer el diagnóstico, puesto que la bilateralidad es un rasgo característico de esta afección.La Neuropatía óptica hereditaria de Leber(NOHL) es una entidad infrecuente y con pronóstico visual sombrío. En pacientes monoculares un alto índice de sospecha es necesario para establecer el diagnóstico, puesto que la bilateralidad es un rasgo característico de esta afección.
Este artículo se publicó originalmente en la Revista MO 2016-01. Autores: Jesica Dimattia, Ezequiel Aranda, Rodrigo Mántaras, Álvaro Suarez Villalobos (Servicio Oftalmología Hospital Provincial del Centenario, Cátedra de Oftalmología, Rosario); Barbosa Sandra y Carmona Sergio (Departamento de Neurooftalmología del Hospital Provincial del Centenario, Rosario). Más información sobre Revista MO en este enlace.
Trabajo
Cuadro: Paciente masculino, ojo único funcional, consulta por primera vez a los 18 años por cuadro de disminución de agudeza visual progresiva de ojo izquierdo (OI) de 10 días de evolución, sin otros síntomas asociados.
Antecedentes: Presentó traumatismo ocular contuso de ojo derecho (OD) a los 8 años de edad, que requirió cirugía por desprendimiento de retina y catarata traumática. Tabaquista de 10 cigarrillos/día.
Examen oftalmológico: Se constató agudeza visual mejor corregida (AVMC) de amaurosis OD y visión bultos OI. Reflejo fotomotor reactivo en OI. A la biomicroscopía del segmento anterior se observaron sinequias pupilares posteriores, iridectomía hora 6, pseudofaquia y opacidad capsular posterior en OD (imagen 1); OI sin alteraciones (imagen 2). En la funduscopía del OI se verificó papila de bordes netos, anillo neurorretinal (ANR) rosado, excavación fisiológica, mácula satisfactoria. OD no valorable por opacidad de medios ópticos. Se solicitó campo visual computarizado en el cual se detectó la presencia de escotoma centrocecal izquierdo.
Diagnósticos diferenciales de la consulta: Neuritis óptica retrobulbar, neuromielitis óptica de Devic, neuropatía óptica hereditaria de Leber, neuropatía óptica tóxico-nutricional, neuropatía óptica compresiva/infiltrativa (tabla 1).
Análisis y estudios requeridos: Se realizaron neuroimágenes de cerebro y médula espinal, laboratorio infeccioso e inmunológico y punción lumbar, que se encontraron dentro de parámetros normales. Electrorretinograma normal OI y ausencia de respuesta en potencial visual evocado por flash.
Se administraron pulsos de corticoides endovenosos, sin recuperación de agudeza visual. Dos meses posteriores al inicio de este tratamiento, la papila óptica izquierda presentó un aspecto atrófico, con ANR pálido y excavación normal.
En 2009, once años después de la primera consulta del paciente (29 años) se decidió realizar el estudio genético para NOHL — no disponible antes——. Este detectó mutación m.11778G>A en el gen MT-ND4 del ADN mitocondrial, por lo que se confirmó la sospecha clínica y se estableció el diagnóstico definitivo de NOHL. Se indicó tratamiento con complejo vitamínico que realizó en forma esporádica y con pobre seguimiento.
Seis años después, el paciente (35 años) retoma la consulta. Se constató estabilidad del cuadro clínico, con AVMC de visión bultos y atrofia papilar izquierda. Se prescribió tratamiento con idebenona a 200mg/día con buena adherencia y respuesta favorable al mismo con mejoría de AVMC.
En la actualidad, el paciente tiene 37 años de edad y continúa su tratamiento con idebenona de 200mg/día. Presenta una AVMC de 0.2 OI con lentes de baja visión, visión cercana de 20/100 OI, reflejo pupilar izquierdo reactivo y, en el fondo de ojo, papila de bordes netos con ANR pálido y excavación fisiológica (imagen 3).
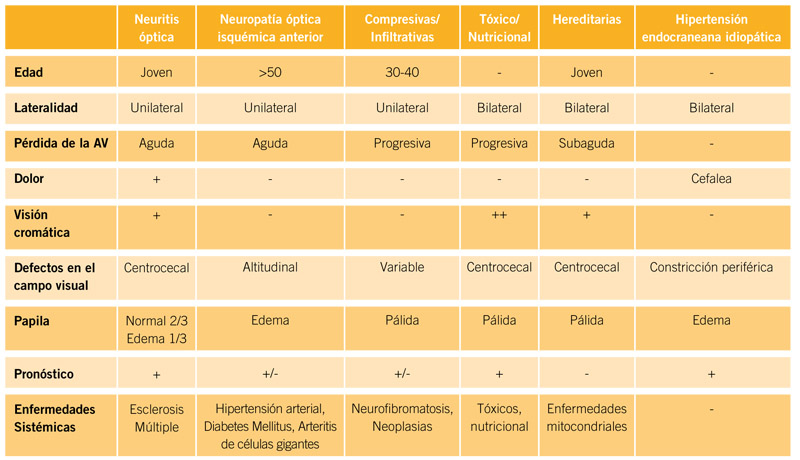
TABLA 1: Diagnósticos diferenciales de neuropatías ópticas.
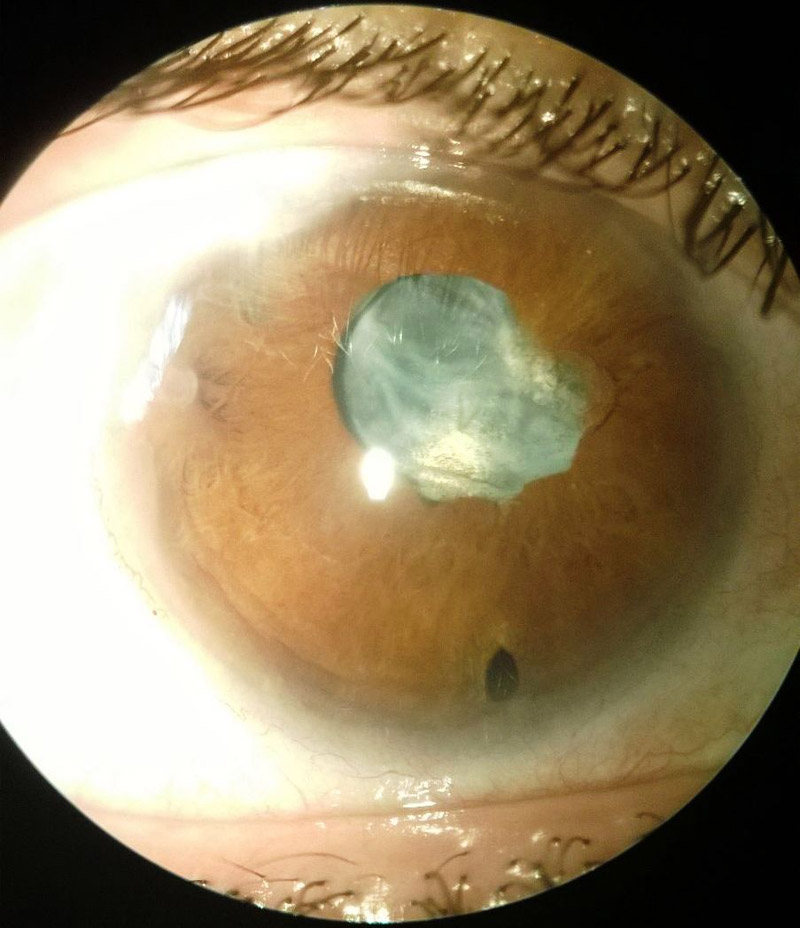
IMAGEN 1: Biomicroscopía del segmento anteerior del ojo derecho.
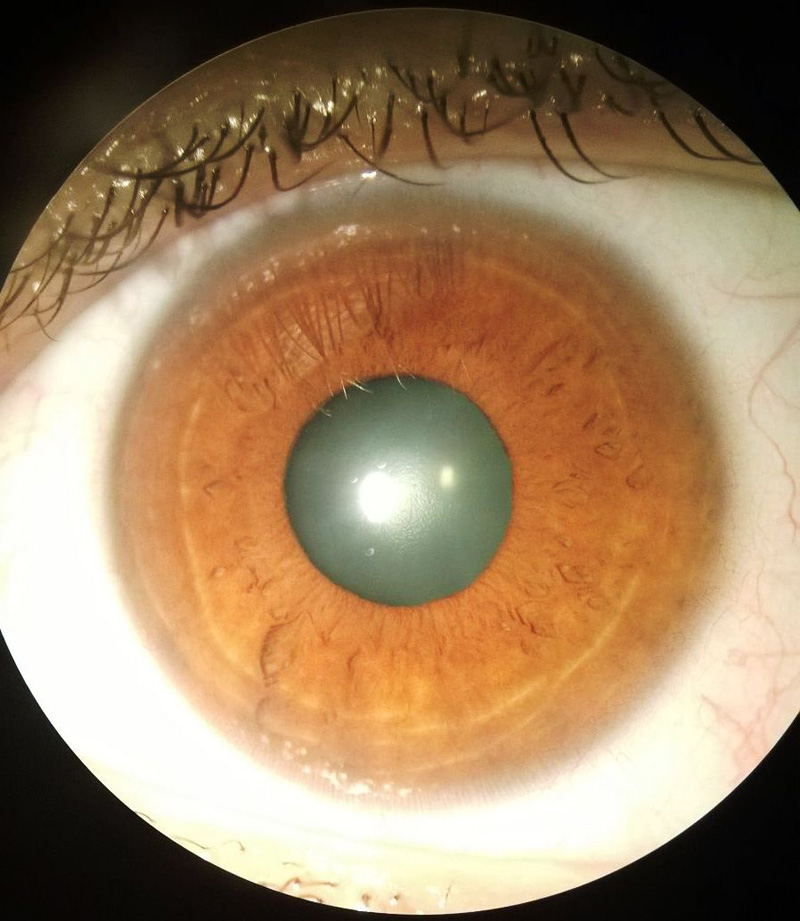
IMAGEN 2: Biomicroscopía del segmento anterior del ojo izquierdo.
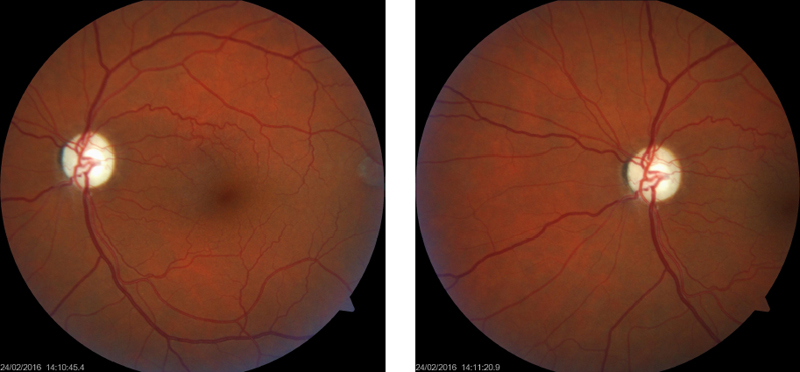
IMAGEN 3: Retinografía ojo izquierdo.
Discusión
La NOHL es una enfermedad causada por mutación en el ADN mitocondrial, de herencia materna y de penetrancia incompleta. El 90% de los individuos con NOHL tiene uno de tres puntos de mutación: m.3460G>A, m.11778>A, o m.14484>C. Hasta el 40% no tiene historia familiar.
Se caracteriza por una disminución de la agudeza visual subaguda, bilateral e indolora.La edad pico para el inicio varía entre la segunda y la tercera década de vida, y el 95% de los afectados pierde la visión antes de los 50 años. La prevalencia es 4 a 5 veces mayor en hombres que en mujeres. Los individuos afectados son asintomáticos hasta que desarrollan visión borrosa con compromiso del campo visual central en un ojo. Síntomas similares aparecen en el otro ojo en promedio de 2 a 3 meses posteriores al inicio. Dentro del año, el 97% de los afectados tiene el involucramiento del segundo ojo. Alrededor del 25% de los casos, debuta con pérdida visual bilateral.
La penetrancia incompleta y el predominio en hombres hacen pensar en una enfermedad multifactorial. Se ha reportado la influencia de factores ambientales, como el alcohol y el tabaco, en el desarrollo de la enfermedad. Algunos reportes vinculan, además el déficit nutricional, la exposición a toxinas industriales, trauma craneano, drogas antivirales y antituberculosas, estrés psicológico o enfermedad aguda con el inicio de la ceguera en NOHL.
La prevalencia de la enfermedad se ha estimado en alrededor de 1 de cada 30.000, mientras que la tasa de portadores de la mutación es estimada en 1 en 350.
La agudeza visual se ve severamente reducida a cuenta dedos —o menos— en la mayoría de los casos, y la evaluación del campo visual muestra un escotoma central o centrocecal denso creciente. Puede presentar además pérdida de la visión de colores y reducción de la sensibilidad al contraste.
Las neuropatías ópticas se caracterizan frecuentemente por anomalías del reflejo pupilar. Sin embargo, en pacientes con neuropatías ópticas de origen mitocondrial, puede haber disociación visual-pupilar tal que, incluso con pérdida visual grave, hay una cierta preservación pupilar.
En la fase aguda se puede observar disco óptico de bordes difusos, edema de la capa de fibras nerviosas peripapilar y cambios vasculares como telangiectasias retinales e incremento de la tortuosidad vascular, aunque el 20% puede no tener cambios en la funduscopía.
Los potenciales visuales evocados pueden ser útiles para confirmar la disfunción del nervio óptico y el patrón de electrorretinograma para corroborar la ausencia de enfermedad retinal.
Las neuroimágenes nos permitirán excluir enfermedades compresivas, infiltrativas e inflamatorias de neuropatía óptica bilateral. LA RMI es usualmente normal, pero puede revelar una señal alta entre los nervios ópticos. Esto último puede representar un edema ligero o gliosis, en la fase aguda o atrófica respectivamente.
El disco óptico se vuelve atrófico dentro de las 6 semanas del inicio de la enfermedad.
Posterior al nadir, la agudeza visual puede mejorar; y esto es más frecuente en los individuos con la mutación m.14484>C. Mejorías significativas en la agudeza visual son raras y la mayoría de las personas presentan una ceguera legal (≤20/200). Sin embargo, algunos factores pronósticos positivos fueron identificados, como una edad temprana de inicio (menor a 20 años), una presentación subaguda con deterioro visual lento y un disco óptico relativamente grande.
Opciones de tratamiento
A pesar de los adelantos científicos, el pilar del tratamiento para la NOHL sigue siendo de soporte más que curativo, ofreciendo ayuda para baja visión y orientando la rehabilitación visual.
Debe recomendarse enérgicamente el abandono del cigarrillo y del consumo de alcohol, evitar la exposición a tóxicos ambientales y el uso de medicaciones con toxicidad mitocondrial.
Varios estudios evaluaron la eficacia de la idebenona, una benzoquinona sintética, en pacientes con NOHL. El estudio LOHN-RHODOS encontró que una dosis de 300mg/x3/día de idebenona durante 6 meses es segura y que los individuos afectados con agudezas visuales discordantes y con riesgo de una mayor pérdida visual en el ojo menos afectado, tienen más probabilidades de beneficiarse del tratamiento con idebenona [Klopstock et al 2011].
En un estudio retrospectivo de 103 individuos con NOHL se demostró que el factor más consistente asociado con la recuperación visual es una iniciación temprana del tratamiento, durante un período prolongado de tiempo, incluso en pacientes con enfermedad establecida y pérdida de agudeza visual severa [Carelli et al 2011].
A pesar de que la idebenona se muestra como un fármaco promisorio en NOHL, se requieren más estudios randomizados, controlados y con un mayor volumen de pacientes para alcanzar un grado de evidencia más alto que permita establecer: efectividad del tratamiento, dosis, período óptimo y qué pacientes tienen más posibilidades de beneficiarse con el tratamiento.
Referencias
- Yu-Wai-Man P, Chinnery PF. Leber Hereditary Optic Neuropathy. 2000 Oct 26 [Updated 2013 Sep 19]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
- T. Klopstock, G. Metz, and P. F. Chinnery, Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy, Brain, 2013 Feb; 135(2): e230.
- Carelli V, La Morgia C, Valentino ML, et al. Idebenone treatment in Leber's hereditary optic neuropathy. Brain. 2011;134:e188.
- L Giordano, S Deceglie, P d’Adamo, et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways, Cell Death and Disease, 2015.
- Rudolph G, Dimitriadis K, Büchner B, et al., Effects of idebenone on color vision in patients with leber hereditary optic neuropathy, J Neuroophthalmol. 2013 Mar;33(1):30-6.
- Moura AL, Nagy BV, La Morgia C, et al. The pupil light reflex in Leber's hereditary optic neuropathy: evidence for preservation of melanopsin-expressing retinal ganglion cells, Invest Ophthalmol Vis Sci. 2013 Jul 2;54(7):4471-7.
- Ayako Mizoguchi, Yuki Hashimoto, and Susumu Ishida, Macular thickness changes in a patient with Leber’s hereditary optic neuropathy, BMC Ophthalmol. 2015; 15:27
- Konstantin Dimitriadis, Miriam Leonhardt, Patrick Yu-Wai-Man, et al. Leber’s hereditary optic neuropathy with late disease onset: clinical and molecular characteristics of 20 patients, Orphanet Journal of Rare Diseases20149:158.
- Meyerson C; Van Stavern G; McClelland C, Leber hereditary optic neuropathy: current perspectives, ClinOphthalmol. 2015; 9:1165-76.
Este artículo se publicó originalmente en la Revista MO 2016-01. Autores: Jesica Dimattia, Ezequiel Aranda, Rodrigo Mántaras, Álvaro Suarez Villalobos (Servicio Oftalmología Hospital Provincial del Centenario, Cátedra de Oftalmología, Rosario); Barbosa Sandra y Carmona Sergio (Departamento de Neurooftalmología del Hospital Provincial del Centenario, Rosario). Más información sobre Revista MO en este enlace.







